▍摘要
2018年,化学界的“AlphaGo”问世,科学家们在《Nature》期刊发文证明AI能够以前所未有的速率进行逆向合成反应,这项新AI工具大大提升科研人员研发新药和其他化合物的效率。在未来,科研机构和制药公司发现新药或者新合成物、新材料的速度将有望达到全新水平。
在AI技术助力化学研究与新药研发的进程下,化学科研仍需大量创新和跳跃性思维,为人工智能提供创新策略,推进人工智能系统不断优化,后者再反过来推动化学研究人员进行更深入的研究。
2020年 6月29日「YOSIA Webinar——人工智能为药物研发和化学研究按下快进键」线上研讨会,汇集化学、制药行业代表及人工智能专家学者,一同探讨化学与制药行业的价值、研究路径及未来发展方向,以期能碰撞出创新的思想之火花。
点击链接观看视频:https://v.qq.com/x/page/k3082h3cxsv.html

▍ 分享嘉宾
主持嘉宾:
· 翁经科,未来论坛青创联盟成员,麻省理工学院生物系副教授、白头生物医学研究所成员
主讲嘉宾:
· 李成涛,Galixir 创始人
· 裴剑锋,北京大学前沿交叉学科研究院特聘研究员,博士生导师
· 申威峰,重庆大学特聘研究员,博士生导师
讨论嘉宾:
· 杨东,西湖大学副研究员
· 杨庆怡,辉瑞制药,医药计算化学科学家
▍ 主题报告
李成涛:AI在化学合成中的应用
人物介绍:李成涛博士是Galixir星药科技创始人。星药科技致力于使用人工智能技术驱动药物发现。李成涛博士主要研究方向包括使用深度学习预测化合物逆合成反应、根据蛋白质序列和三维结构以及化合物性质等对化合物进行筛选等多个药物发现领域中的重要环节,在人工智能届顶级会议上发表了多篇论文。李成涛本科毕业于清华大学姚班,并在麻省理工学院取得了计算机科学的博士学位。
合成以及逆合成是有机化学中的一个重要命题。逆合成旨在寻找能够合成目标化合物的反应物和合成路径。
通过运用深度学习技术进行基于化学反应数据的学习,人工智能可以帮助化学家设计合成路线并且选择中间体化合物,从而极大地减少设计需要的时间。
这对于药物的研发、围绕着药物和中间体化合物进行的专利保护、以及提高药物的生产效率都有着重大的意义。非常高兴和大家分享人工智能在生物医药特别是化学合成领域的应用。
为什么做化学合成呢?因为化学合成在整个生物医药和制药领域都扮演了非常重要的角色。生物医药和制药研发流程大概分为两个阶段:第一阶段是临床前阶段,涉及到选择蛋白质靶点、选择设计先导化合物(包括先导化合物的优化、筛选)。
设计出好的分子之后,再进行体外和体内实验,最终如果实验结果不错,我们可以拿到临床许可进入临床阶段。第二阶段是临床阶段,就是临床I、II、III期,再之后就是获批上市。
其实这两个阶段都有很长的研发时间。尤其是临床前阶段,大概有5到8年的时间。现在研发一款药物,从靶点选择到获批上市的时间平均是14年,这是一个非常漫长的过程。漫长的时间意味着巨大的投入。从70年代开始,总资金投入大约每十年翻一番,这被总结成一个比较著名的定律,叫做Eroom定律,Eroom是摩尔定律的反写。
Eroom定律的意思是,随着时间的推移和技术的发展,新药研发的效率反而越来越低,并伴随着投入越来越高。现在研发一款药,平均要花费20亿美金以上,这是非常巨大的投入。同时新药研发伴随着巨大风险,而且,随着时间的推移越来越高,即研发成功率是非常低的。
高昂的价格和过长的研发周期造成研发投入的回报率逐年降低,在2010年是10%,到2019年小于2%,现在则可能更低。其实世界上有很多种疾病,但是真正有治疗方法的可能不到一半,有很多疾病并没有被关注,因为研发成本实在太高了。
人们一直在思考如何降本增效,人工智能作为可行的方案出现了。人工智能近年迎来了爆发式的发展,从数据、算力再到算法都有了突破性的进展。AlphaGo是下围棋的AI,人们好奇研发这种算法有什么用?其实围棋和化学合成之间有非常强的联系。围棋每一步都有数十至上百种可能性,每一步棋都会对后面的策略造成很大的影响。
围棋是最终目标是赢,但每一步都有数十上百种可能性,要走很多步才能知道是赢还是输,这造成了巨大的搜索空间,用计算机去直接搜索所有可能策略是做不到的。DeepMind首先也用了很多算力,但是他们的算法很先进,可以快速找到赢面更大的策略。

这和化学合成有什么关系呢?首先讲什么是化学逆合成反应。当我们设计出目标分子后,逐渐把目标分子分解成中间体化合物,再分解成构造快,这样其实就找到了一个目标分子的合成路径。这就叫逆合成反应。
逆合成反应其实也面临和围棋类似的问题。
比如,我们要合成分子1,可能用分子2和分子6合成,也可能用分子8和分子9合成。分子8能够买到,或者结构相对比较简单,我们知道怎么合成它,但是分子9不是很好合成。分子6和分子2都可以买到。于是相比之下我要找分子2和分子6的合成路径。
分子2可以由分子7和分子8合成,也可以由分子3合成。用分子3合成相对比较简单。于是我就找分子3的合成路径,发现分子3可以分解成分子4和分子5,分子4和分子5都可以买到。那么我们的目的就达到了。综合来看,逆合成反应就是不断地把现有的不能合成或者不能买到的分子,分解成一系列的前体,使得每一个前体都可以买到或合成。
简化来看的话,比如合成分子A有两条路可选,我选择其中一条,合成分子B也有两条路可选,我选择其中一条。这其实是相对比较简单的情况,三步就完成了,每一次最多从二选一,即使要把所有路径都试一遍,也是比较简单的。但现实生活中往往不是这样,合成A的方式很多种,合成B和D的方式又有很多种,这跟AlphaGo倒是挺相似的。
这个过程也涉及到巨大的搜索空间,造成了计算上的复杂度。所以我们需要借鉴AlphaGo的网络结构和算法,包括它的计算方式。
我们在2018年的一篇Nature论文中看到了解决这一问题的曙光,这项研究相当于把整个的化学逆反应合成分成两个模块。第一个模块是单步逆反应预测,每一种化合物都有数十种合成方式,但是这数十种合成方式可能是不知道的,这跟围棋不太一样。我们需要一个新的模型来预测分子的前体。第二个模块是多步逆反应搜索,这借鉴了AlphaGo的蒙特卡洛树搜索方法,计算机测试化学实验达到了非常好的效果。
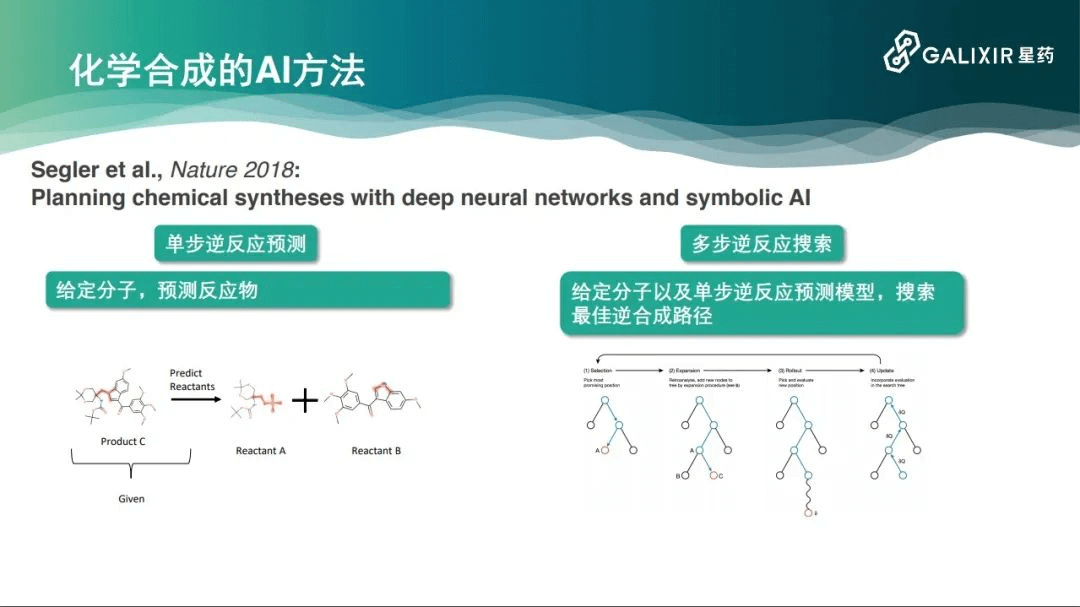
此外,还有一些方法的第一个模块是基于序列预测的。一个化学分子在计算机里可以表示成一个字符串。要从一个化学分子预测它的前体,这本身就是一个字符串到字符串的映射,可以用自然语言处理的方法解决。
对于中等难度的分子,比较有经验的化学家大约几个小时到一天可以做出来相对比较好的反应路径,但是人工智能可以把整个过程加速到秒级别。以上所讲的其实只是整个药物研发中的一小块,当然逆反应合成是整个拼图中非常重要的一块。如果希望做到端到端的药物研发的话,需要很多其他模块,比如人工智能在靶点筛选、药物设计、药物分子生成、药物筛选中其实也可以发挥很大作用。
裴剑锋:弱AI时代的药物设计
人物介绍:裴剑锋,北京大学前沿交叉学科研究院特聘研究员, 博士生导师,在PNAS, JACS, NAR, Nature,Chemical Science等国际著名期刊上发表论文50多篇,兼任中国化学会计算机化学专业委员会委员,中国药理学会定量药理学专业委员会委员, 中国医药生物技术协会生物信息学分会委员,中国民族医药学会信息与大数据分会理事。主持国家自然科学基金,国家科技重大专项,科技部重点研发专项等科研项目6项,在国内率先开始人工智能化学信息学和药物设计研究, 曾获得中国药学会施维雅青年药物化学奖,中国化学会青年计算化学家奖以及药明康德生命化学研究奖。
我首先介绍人工智能技术的原理和应用场景,然后介绍机器学习的分类及发展历程。整个报告的重点是介绍人工智能技术在药物设计及化学信息学中的应用及研究进展,尤其是多种深度学习技术的核心思想,并展望未来科研模型将会如何转变。
机器学习是属于人工智能中的一类技术,其包含了监督学习、非监督学习及强化学习。而深度学习又是隶属于机器学习领域中的一种最新技术,其在大数据方面表现出了更好的性能,但也更依赖大数据。

其实,人工智能朝两个方向发展,一个方向是仿生学,一个是工程学。目前,人工智能在视觉计算、语音识别、自然语言处理方面都表现出了卓越的性能。但是,这种感知智能仍然是一种弱人工智能,通常只能做一种事情,无法实现认知智能,这方面还有很多工作可以做。
回到药物研发,传统的基于实验的高通量筛选化合物,效率比较低而且风险很大。引入人工智能的目的就是在于提高药物分子设计的效率和成功率、提前预知错误的药物分子或合成路线,及早止损。目前,人工智能主要应用在药物发现阶段,在其他的阶段也有些应用。但是,回顾以往的研究,迄今为止还没有完全证实人工智能技术能在已成功的新药研发中发挥了绝对作用。下面介绍我们的一些基础性工作。
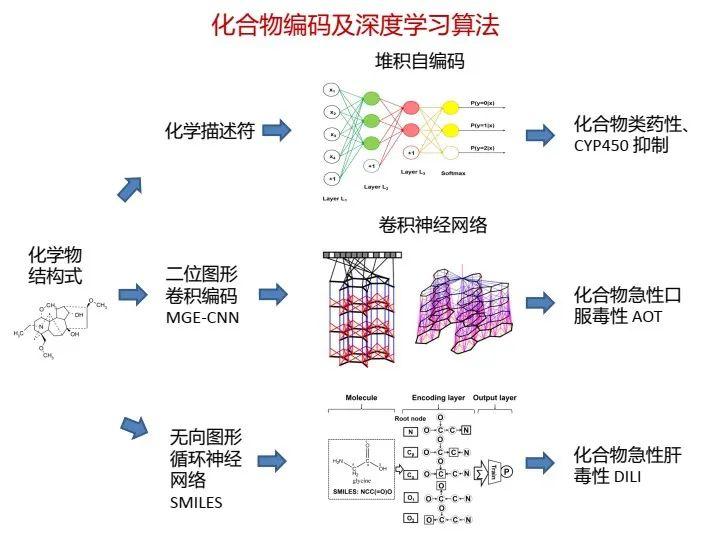
在基于人工智能的药物研发中,药物化合物的分子结构编码是必须解决的问题,一般包含三种方法:一是采用大量的化学描述符(descriptor),每个描述符描述分子的各种局部特征或有限的化学信息,加以组合形成矢量便可以提供化合物较为完善的信息;二是采用类似图卷积的思想,将不同半径范围内的分子子结构特征提取出来,形成分子指纹,例如类似于ECFP4环形分子指纹;三是采用自然语言处理技术,直接处理表示分子结构的线性字符串。
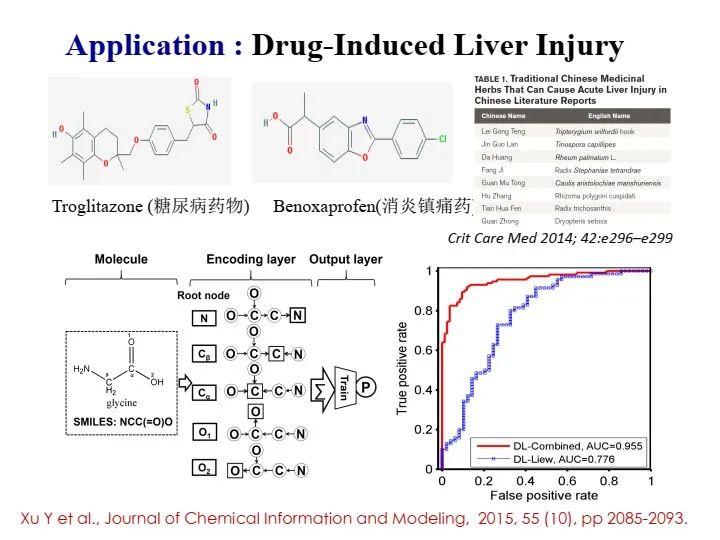
例如,我们将第三种方法应用在肝损伤的预测上。
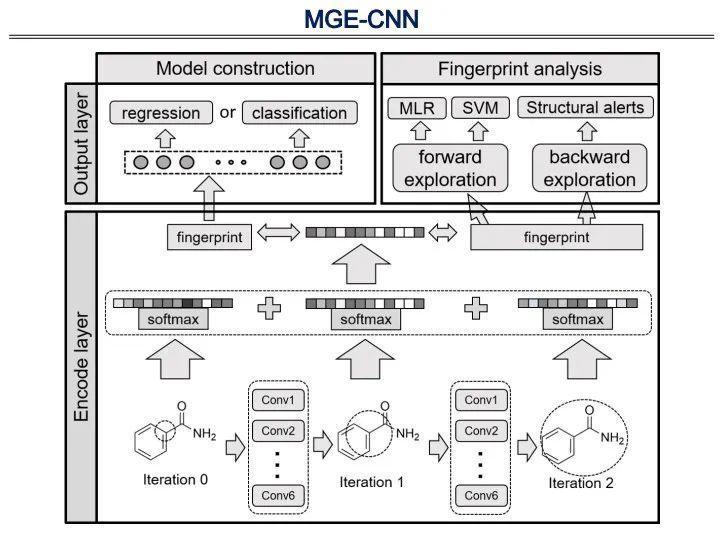
这是我们运用第二种方法,在不同的分子半径上提取特征,完成构效关系建模及分子指纹分析的工作。
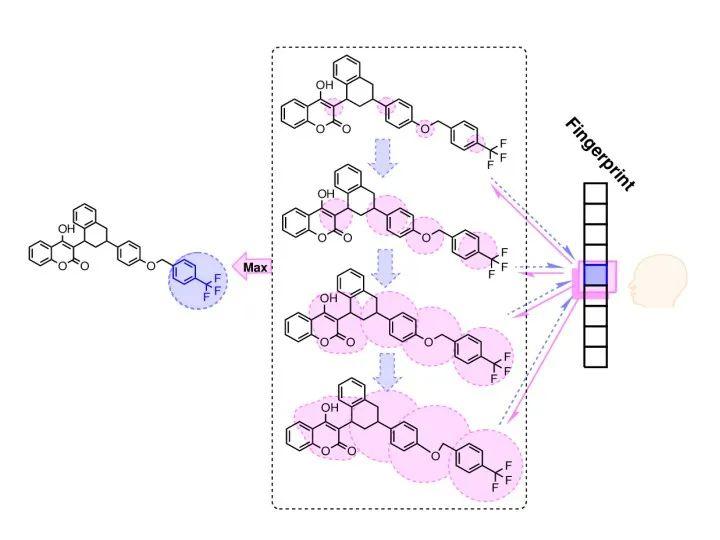
卷积神经网络的特点就是可以提取出图像的局部特征,如果应用在分子结构上,也可以找到一些与目标性质关联的局部特征,从而提供一些解释性证据。下面也提一下图结构及图神经网络,也有不少工作。
由于分子结构可以看成无向图,所以可以利用图神经网络来学习,但是也要解决一些其中关键的问题。在实现分子结构的编码及相关的性质/活性预测后,还需回答如何生成目标分子结构的问题。
这里讲一下,现在非常热门的生成模型,也可以用在药物分子生成上,可以通过循环神经网络(Seq2Seq)来实现,也可以通过变分自编码器(VAE)或者对抗神经网络(GAN)来实现。
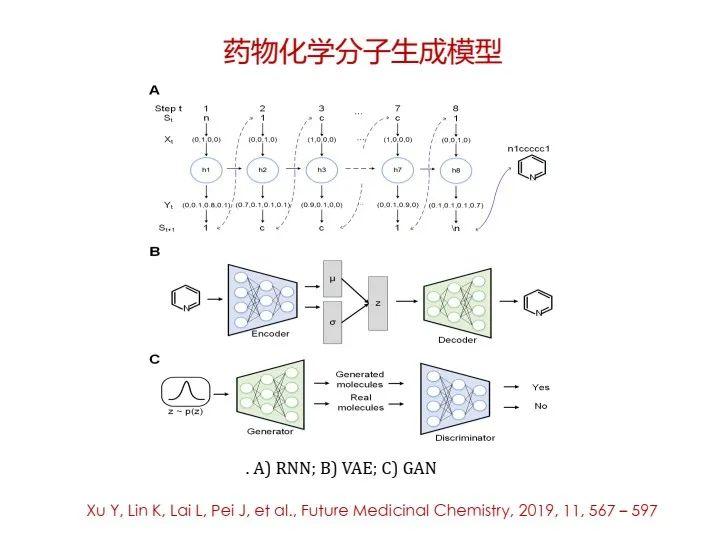
但是,这种思路没有考虑药物分子与靶点的结合能力,这是受限于分子结构二维表达或线性字符编码。我觉得未来应该向三维的分子生成方式发展,这样可以引入一些分子三维结构上的组装可行性约束。我们正在努力实现三维分子生成,同时也做了一些合成路径设计方面的工作。
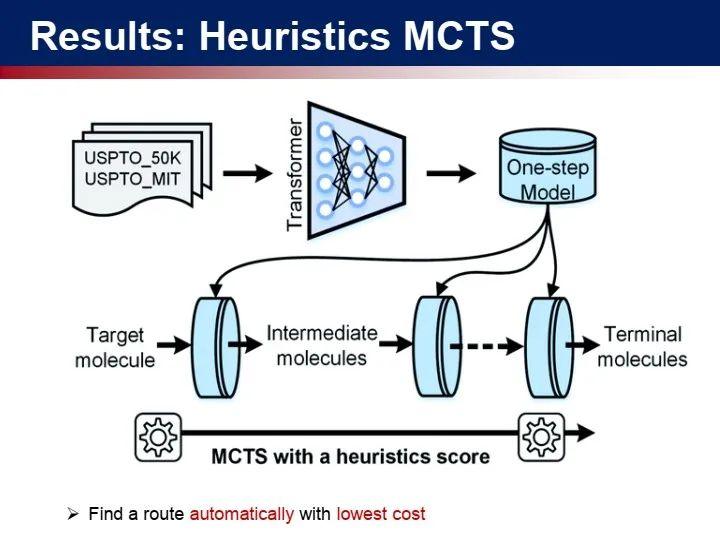
我们发现如何对合成路径进行打分是一个很难的问题,即怎么定义一个合理的基准来评价最佳的反应路径。
但是,现有大多数研究提出的模型只能在基准测试数据集上进行评估,离实用还有一定的距离。我们期望能够开发更加实用的人工智能药物设计工具,通过把很多工作步骤串联在一起,改善模型的实验可验证性。因此,科研分成三类:
1)开普勒模式:从数据中总结规律,典型的方法就是机器学习
2)牛顿模式:先把公式从原理上提出来,然后模拟真实体系。但量子力学很难处理复杂的大分子问题。
3)用人工智能、机器学习和物理模型结合的模式:无论在材料设计方面,还是在药物设计方面,我觉得都是一个很好的新模式,既可以面对复杂问题,又不脱离基本原理的支撑。这可能导致未来科研模式的较大转变,会引发一些颠覆性的创新。
申威峰:AI在环境健康安全溶剂开发中的应用
人物介绍:申威峰,重庆大学特聘研究员,博士生导师,重庆市青年拔尖人才。国家工信部工业节能行业标准化总体组专家委员;中国工业节能与清洁生产协会专家委员会委员;《中国化学工程学报》青年编委;Frontiers in Chemical Engineering副主编;中国系统工程学会过程系统工程专业委员会委员;世界绿色智能制造技术进展大会国际专业委员会委员;中国化工学会化工过程强化委员会委员。2009年在重庆大学获得硕士学位,2009-2012年在法国图卢兹大学国立综合理工学院获得工学博士学位,2012-2015年在荷兰壳牌石油集团/美国克拉克森大学担任助理研究员。作为负责人主持国家级、省部级、中央高校等项目10余项。主要从事人工智能在绿色溶剂开发和化工过程强化中的应用。发表SCI论文60余篇,申请发明专利9项。
随着全球污染问题的日益严峻和环保意识的不断加强,减少溶剂的使用量或开发环境友好型溶剂是解决化工环境安全问题的重要举措。数据驱动的分子设计技术不仅可以高效便捷地筛选候选溶剂,还能够综合考虑溶剂分离性能、经济效益、以及环境健康安全(EHS)等潜在影响。
基于人工智能技术的深度学习神经网络,采用高性能计算平台进行智能识别和提取分子结构的特征,捕获微观分子结构与物质宏观性质之间的关系,运用人工智能技术建立更智能化的定量结构性质关系预测模型,有效地预测所需溶剂和化工产品的环境健康安全性质,推动绿色化工产品与可持续化工过程的不断开发。今天和大家分享我们团队最近基于人工智能技术对考虑环境健康安全潜在影响等多方面性质的溶剂开发的最新研究进展。我们的研究方向和李成涛老师相反,他做的是逆向过程,我们做的是正向过程。
我从研究背景、研究思路和方法、研究案例以及结论和展望这四个方面展开介绍。在石油、医药、化工、环境、食品等行业,溶剂是反应、分离等过程最常用的物质。据Nature的一篇文章报道,化工分离过程就要消耗世界能源的45%到55%。另一方面,这些行业在生产过程也会产生大量有机废液,我国每年产生有机废液量在35万吨以上,不少企业因为废液污染面临责令整改或者搬迁。如何升级换代,如何开发绿色溶剂对企业来说有重要意义。
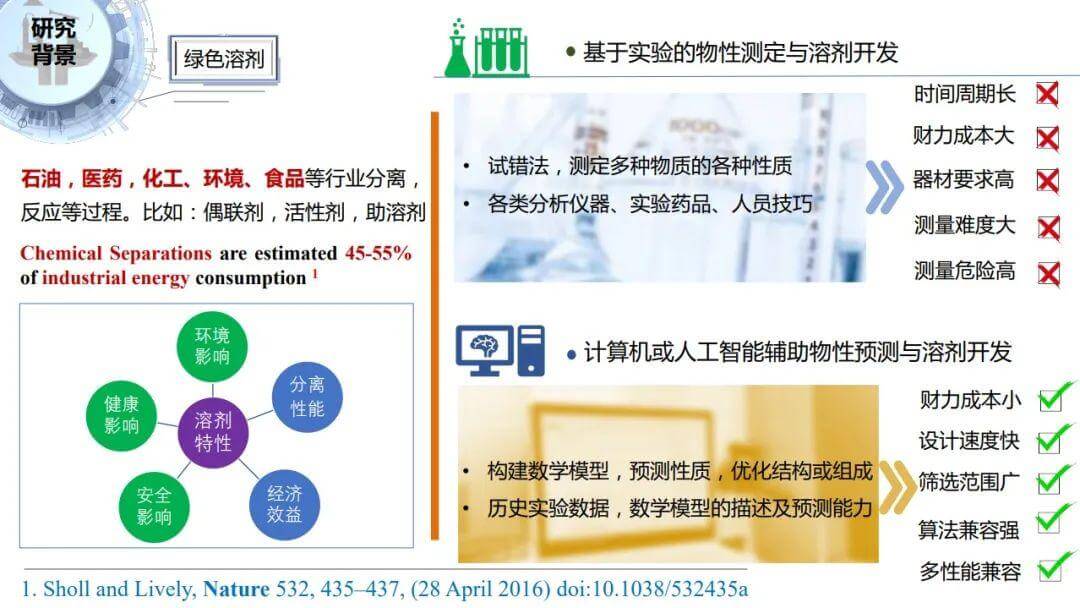
传统的实验试错的方法,使得对物性的测定和溶剂的开发不仅周期非常长,而且耗费大量人力物力。同时可能有些物质难溶或者不溶,导致测量难度非常大。
有些物质毒性非常大,导致实验过程非常危险。这些问题都不利于实验的大范围广泛开展。所以人工智能辅助筛选设计开发技术可以实现便捷、快速的筛选溶剂的同时,考虑环境健康安全潜在影响的多种性能,而且筛选范围更广。
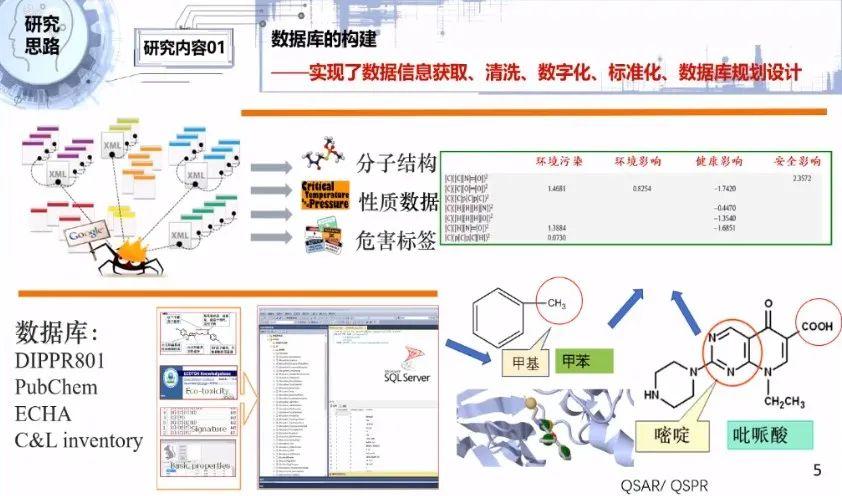
我们的研究过程从四个方面展开,包括数据收集、数据整理和特征提取、模型搭建、模型训练与评估。智能化溶剂开发的首要任务是构建一个数据库。我们通过不同的途径开发了一些软件,通过不同的数据库获取到各方面的数据信息,包括分子结构信息、基本性质数据、环境健康安全数据,以及构效关系、QSAR、QSPR数据。
构效关系是我们研究的重要问题。比如甲苯里面带有甲基,吡哌酸里带有嘧啶,不同的分子结构因为带有不同的基团类型或者数量不同,会导致分子具有不同的性质,这种结构影响性质的关系就叫构效关系。我们对这些对应关系建立了构效关系的系数矩阵数据库。
我们对收集的数据在后期进行了清洗、数字化、标准化,采用SQL Server来规划设计数据库,建立了一系列的物质识别、基础物性、结构信息、毒性数据、安全性数据等一系列的不同类型的数据的收集表。这是前期数据库的工作。
我们之后还搭建了深度学习架构,具体包括分子结构特征的提取、构建深度学习网络模型、训练和预测等工作。
总体思路是,第一步把分子结构转换为非环有向图的结构,目的是让计算机能够遍历整个分子图。第二步将非环有向图的每个顶点定向量化。第三步通过训练,将分子结构进行向量化。第四步通过搭建的深度学习算法对构效关系进行建模,对目标分子结构向量和目标性质进行关联,最后输出一个我们预期的预测目标性能。
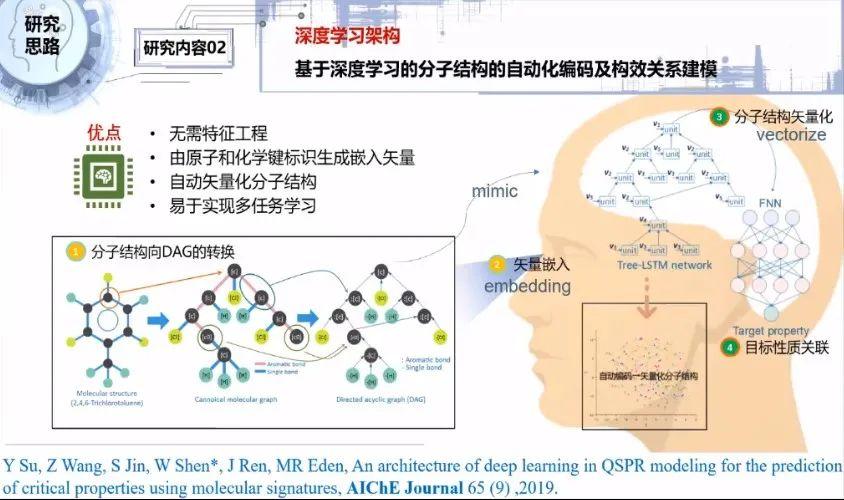
以下对研究方案作更详细的介绍。第一步,首先对分子结构进行前期处理。从分子结构的任意一个原子出发,依次从左右两边展开,在另一个闭合碳原子打开,把打开的原子赋予标识碳的元素符号C_0,这个分子就被打开成一个类似于神经网络的树状结构,使得神经网络方便处理。
这从侧面说明分子的化学结构和计算机领域的神经网络是非常相似相通的。另外,化学键的信息没有保留到结构信息里面,所以要把化学键以字符形式表示,合并到每个节点。
通过自然语言处理技术,把每个顶点的标识转化为嵌入向量标识。基于树形长短记忆神经网络对新的分子结构进行遍历,或者说搜索定位,按顶点位置搜索对应的嵌入向量,这样可以保留每个原子和化学键的信息。当我们把树形长短记忆网络和单个的前馈神经网络进行联合时发现,它可以完成单任务构效关系的智能建模,并且可以预测单目标性质。
当我们用树形长短记忆网络与多个前馈神经网络进行联合时,可以完成多个任务的QSPR建模,同时可以完成多个任务的构效建模,这样一个模型就可以预测多个目标性质。我们分别从基础物性、环境性质、安全性质、EHS潜在风险评估深度学习建模方法的有效性,下面分别对案例进行介绍。
案例一:临界性质模型我们可以在训练集、测试集取得非常好的训练效果。尤其是对同分异构体的识别,深度学习模型表现出比其他模型更好的识别能力。
案例二:环境性质模型我们验证了模型在更大规模数据的有效性,在这种情况下取得了更好的预测效果。并且我们与现有的基团贡献的模型进行对比,发现深度学习模型表现出了更好的性能。
案例三:安全性质模型研究闪点、自燃点、燃烧上下限性质,来验证多任务学习模型的有效性。结果发现,我们开发的模型在同时输出四个性质的时候,均能表现出非常好的预测效果。我们还做了基于机器学习的溶剂的环境健康安全潜在风险评估。我们采用数据库里的数据性质,按照一定规则对各种性质健康风险进行评分,最后将多个物质的风险评分与分子指纹作为数据样本,通过机器学习算法进行关联,最后形成了环境健康安全风险评估模型。
通过评分模型的预测值和实际的混淆矩阵可以看出,训练集和测试集大部分数据集中在中间对角线上,这充分说明我们可以得到一个理想的训练集的预测精准度。通过总体加权,平均统计发现,这些准确度可以达到90%左右。
通过这项研究,我们总结出在环境、健康、安全各方面性质对应的分子结构的高风险分子结构清单。总结一下,我们的研究工作主要体现在四个方面。
第一,开发了分子结构的改进编码策略,结合词嵌入算法及树形长短记忆神经网络,可以使计算机自动向量化分子结构信息。
第二,通过构建的深度学习架构,实现构效关系的智能建模,获得模型的较好预测能力。
第三,基于分子指纹和机器学习算法的构效关系建模,可以在评估溶剂的时候不依赖性质数据,筛选出对于健康、对于安全相对应的高风险的分子结构特征。第四,我们开发的技术可以同时考虑功能、经济效益和环境健康安全各方面影响的性质。

在未来,我们将进一步研究深度学习构建关系的应用化学空间和预测能力,研究模型的不确定性。同时要进一步研究逆向分子结构的设计策略。我们还将进一步把深度学习框架推广到药物、材料、萃取溶剂等其他方面物质的预测、分类或者开发。
▍ 跨学科讨论
人工智能如何在制药和化学领域发挥作用
翁经科:下面讨论人工智能在制药化学的运用,以及能帮助解决哪些传统手段不能解决或者很难解决的问题。
杨庆怡:说到传统计算方法,我们一般会联想到量子力学计算方法或者分子动力学等方面的模拟计算方法。相对于经典的量子力学计算方法来说,AI模型能够覆盖更多的数据,从更多的数据里面获取信息,传统科学计算很难捕捉到这些信息,这是AI很大的优势。
比如上面提到的,深度学习模型能够通过对大数据的训练和学习提供更好的预测以及产生新的分子结构,这是以前经典的科学计算不能做到的。
杨东:现阶段AI的发展带来的最大帮助还是在新药研发,新药研发是一个系统工程,包括了很多环节。这些环节实际上都是非常耗时耗力的,以前都是以低效的方式进行,成本也非常高。一项新药的研发费用高、研发周期长、研发成功率低。随着AI技术的应用以及不断发展和成熟,可以为新药研发减负。值得一提的是,AI技术在药物靶点的设计上也有应用,可以利用自然语言处理技术让AI阅读海量文献,总结科学家的研究成果,来设计被人们忽略的药物靶点。
李成涛:我了解到科学计算由于计算复杂度的限制,所以计算过程中都会应用近似,这些近似可能会引入系统性的误差,这种误差很多都是没有办法除去的。相对于传统的计算方法,AI在数据量足够多的情况下,某种程度上可以减少或者去除系统性误差。这一现象有相关的理论支持,即当数据量足够的情况下深度学习网络可以拟合任意一个函数。从AI角度来讲,最核心的问题是研发规模。之前研发一款药可能几百个人一起讨论,可能最后设计出成百上千种分子。AI使我们能一次性模拟几百万种药物。只要模型是合适的,完全可以解决规模的问题。
AI在化学和制药领域的应用瓶颈
翁经科:第二个讨论议题,人工智能在现代制药和化学领域的应用瓶颈在哪里?
裴剑锋:第一个瓶颈是数据问题。在药物研发应用场景下,数据很难自动产生。比如说,做天然产物的合成,数据不够,可能需要人来打标签,那可能十几轮跌代后就做不下去了,如果用模型来生成数据,精度和准度也是有限制的。另外,在药物研发里,很多数据没有阴性数据,但是作为机器学习的模型,负样本非常重要。没有这个负样本,数据就不平衡。这个问题始终存在,需要很大的数据系统来支持。
第二个瓶颈在于人工智能本身。药物研发就是人工智能的一个应用,也存在一些局限。因为药物系统本身很复杂,将一个前沿的东西应用到复杂系统,这里面就会出现瓶颈。比如药物专家的经验的学习和传承,人工智能很难处理。第三个瓶颈是人才,因为这是很强的交叉学科。
申威峰:在化工数据中,比如环境方面的物性数据是非常难收集的,这也是制约模型预测能力的非常重要因素。我们团队大部分工作都在做数据收集,数据收集的好坏直接影响模型预测能力。刚才裴剑锋教授提到药物的无效数据不好获取,但是化工厂内无效数据很多,因为我们化工厂一般都有一个DCS自动控制系统,它可以把所有数据都采集下来,但是有一个问题是很多数据是重复的、无效的,处理工作量非常大。
另一个比较大的问题是,人工智能技术大多是基于概率模型的,所以对一些现象和结果不能提供充分解释。所以无论制药行业还是化学领域,都有一定的特有知识、机理或者机制为标准,并不是所有问题都可以单纯用人工智能技术来解决。如何将基于人工智能技术的黑箱模型与化工领域的白箱模型(比如能量守恒、物料守恒等)结合,形成一套完善的研究方法,是目前化学和制药领域都面临的难题。
交叉学科的成才之路
翁经科:第三个讨论议题是人才问题,各位嘉宾走到今天进入了人工智能和化学制药领域,肯定都有自己独特的故事,所以想请大家分享一下,你们是怎么样进入到现在的职业轨道的?
杨庆怡:我的职业轨道比较简单,主要是幸运。我本科毕业以后就到美国读博士,博士毕业后就在GSK制药公司工作几年,然后来到辉瑞,一直从事计算化学工作。我们团队对AI非常感兴趣,也投入了很多研究,想发掘AI在制药上的潜力。
杨东:我本身是做生物信息学的,我的导师的课题组主要是做癌症研究。癌症非常复杂,每个人的肿瘤几乎完全不相同的,因为携带有不同的基因突变。科学家近几十年针对这些不同的突变开发出了上百种抗癌药物,但是癌症对症下药的问题到现在也不令人满意。
传统的办法是利用生物标记物判断病人是否适合某种药物。到现在为止,这个效果也不是特别令人满意。我最早接触到人工智能,是2016年AlphaGo战胜世界冠军的时候。
我发现AlphaGo能处理非常复杂的模式,当时就想可以通过人工智能的办法学习人类不同肿瘤基因组,实现对肿瘤患者的个性化用药。
于是我开始学习人工智能的知识,也开始写代码来实现人工智能的算法。研究过程中我发现光有病人肿瘤信息是不够的,还要把抗癌药物化合物的信息也融入进来。所以我又想办法学习了化学,把抗癌药物化合物的信息整合收入到人工智能体系里面,希望药效预测能做得更加准确。
所以我为了自己科研的目标,学习了不是自己主专业的知识。我一开始觉得人工智能离生物特别远,其实当真正投入到科研当中,感受到想要实现一个目标的时候,就不要考虑太多,就把自己的精力专注在知识里,哪怕不是这个领域的,认真学习还是能够慢慢的掌握。并且AI现在还是起步阶段,没有特别多知识需要积累。我建议大家不需要害怕,大胆去尝试,认真读文献,亲自做实现,最终还是能达到自己研究的目标。
李成涛:我从本科到博士一直在从事人工智能领域的研究,在博士期间萌生了创业的思想,后来才转向人工智能和化学这个交叉领域,然后创立了星药科技。我本科一直在学计算机,化学基础有限。为了多了解领域知识,上过课,看过书,现在对人工智能+新药研发领域有了对框架的整体了解。
2019年初,我提前毕业然后做了这家公司,到现在也就摸爬滚打了一年半,还在不断学习和进步中。作为原本是人工智能专业的人,要进入化学领域或者制药领域其实是有一定门槛的,当时花了很多时间,但我觉得这件事情确实很有意义也很有意思。就我个人经历而言,假如你是做AI的人,对其它领域感兴趣,不妨多花一些时间了解,坚持去做。
申威峰:我过去的科研方向是计算机辅助化工生产,对化工生产过程进行模拟、优化和控制,这和人工智能还是比较接近的。2016年回国后,我们团队开始进入人工智能方向。化工行业很多高端产品需要国外进口,因为国内的技术比国外差很多。但是现在我们可以通过更智能的工具来辅助精准决策,从而提高产品质量。
关于化工领域的人才培养,这个方向是交叉学科,需要同时学习两个学科的知识和技能。我呼吁国内高校开办相应的交叉专业,目前有人工智能专业兴起,但是交叉的专业还没有,所以开设交叉专业,配置相应的课程和教学计划。在学生学习化学知识的同时,可以加强数学理论、化学信息、软件开发相关课程的学习,这样才可以适应未来对交叉学科领域人才的需求。
裴剑锋:我是学生物出身,当时比较喜欢计算机,自学了很多计算机的知识,博士的时候转到计算机辅助设计,我一直在这个方向上走。关于交叉人才培养,有药学和化学基础的可以多学习一些人工智能的知识。关于交叉人才培养,无论是学药学、学化学,可以先在专业方向打好基础,再学习人工智能知识。
北大前沿学科研究院是国内第一家专业以交叉学科为主的研究院,注重培养各方面的交叉人才。如果想进入这个行业,希望同学们多开发一些底层的算法或者代码,少在别人基础上发展,这对于中国的学科发展非常重要。
制药、化学和AI的未来十年
翁经科:我们现在进入第四个讨论议题,请各位预测,十年以后制药、化学、人工智能的未来发展前景。
杨庆怡:未来很重要的一个发展方向是数据变得越来越多,化学数据库平台可以给化学家和生物化学家提供很大的便利。在化学信息学的基础上,如果AI技术能够从信息里面探索出真正的知识,更一步将数据变成知识,相信能够带来更大的突破。比如,更准确地预测分子结构,更高效率地找到初始小分子候选清单,这对小分子和靶点识别的研发非常有用。
杨东:现在化学新药的研发已经开始运用计算机的算法来辅助药物设计进行虚拟的筛选模式。目前可供计算的范围或者预测能力还比较有限,还是要大量依靠人工做实验去验证。
十年以后,很可能人们还是以计算机结合实验验证的模式进行,唯一不同是人工智能技术的不断成熟以及数据的积累,二者的占比会发生很大变化。可能未来进行大量模拟计算预测将成为主要部分,包括靶点发现、药物筛选、优化都是由AI来完成。经过若干轮的严格模拟筛选,只剩下很少的分子,我们才会人工做实验来合成,然后验证安全性和有效性。
李成涛:十年之后,人工智能的发展主要在三个方面,分别是算法、数据和算力。算法就不用说了,也很难预测,但是相信会有长足发展。第二是数据,我们现在有很多方式积累数据,比如实验室做的实验数据可以作为积累,当然也有高通量的实验平台,这也为积累数据提供了便利。
数据量足够之后,机器学习模型可以拟合到和自然更贴切,这样输出结果相对就会更好。第三是算力,尤其是专有芯片。比如说,现在摩尔定律暂时失效了,但是专有芯片发展还是很快的。相信十年之后人工智能会对整个制药和化学领域产生非常深远影响,具体就体现它能算的更快、规模更大、算的更准。
裴剑锋:如果General Intelligence未来十年发展的很好的话,场景就非常乐观。比如在计算化学和化学机制的预测上,可以看到机器学习,量化和密度泛函理论结合的很好,数据量足够的时候,计算精度已经接近那个精度了,并且它的速度很快。
如果采用不断迭代的方法,也许我们可以推动量化计算或者模拟它的体系的增大,对理论化学,这是非常有用的。另外,可能更多的数据大家会去注意积累,中国要成立国家数据中心,这也是非常重要的。可能十年后真的会产生一些比较大的突破。
General的东西,我最希望它在自然语言处理上产生巨大突破,这样真的是能够帮我们读文献,把这么多散乱的非结构化的信息,组织成有用的知识。这无论对哪个行业,对制药行业都是非常重要。
申威峰:以后基于人工智能的化学产品的开发软件或者是化工辅助决策软件会大量出现,科技工作者可以利用这些工具快速筛选想要的分子结构、产品配方、合成路线等,大大提高合成效率,保证高纯度产品。
同时,做化学合成或者药物合成的一些重复劳动力的科研工作者可能会减少,并将人才转移到从事人工智能和化工药物结合的方向上。人工智能技术不仅可以加速研究的进程,同时可以向着更绿色、更健康、更便捷的方向发展。
AI如何和制药、化工产业对接
翁经科:最后的讨论议题是,人工智能如何和制药、化工产业对接,以及学术界如何与工业界对接?
申威峰:对于化工方向,首先可以开发人工智能辅助的软件,帮助企业合成和分离过程的决策。另一方面,化工是一个质控系统,可以借助人工智能和化工制造系统结合,形成自优化、自诊断的智慧化工系统。
我们要基于人工智能大数据、物联网,化工厂的设备监测技术融合,来构建一个智慧化工决策系统,开发能够分析化工厂企业的在线数据,开发适用于物联网的传感器,开发质控系统和人工智能的交互平台,形成决策软件包,这样可以帮助企业在化工过程中化工厂里面优化生产效益、减少能耗,控制生产风险,同时提高产品质量,我认为可以通过这种方式来对接。
李成涛:关于人工智能如何和现有的制药或者化工产业对接,分两个方面,第一个方面从人工智能角度来讲,很多做人工智能的毕竟不是出身于药物研发或者化工产业,所以他对化工产业、药物研发的了解相对有限,需要多交流。比如说产业的痛点,人工智能能解决的问题,能提供什么产品,这对于企业非常重要。另外,从制药和化工产业角度来讲,多交流也非常有益。现在对人工智能了解相对比较少的领域专家,他们可能对其期待不高或不切实际。
杨东:人工智能现在存在可解释性问题,这个问题涉及医药、健康领域时,人们往往会比较谨慎。当人工智能预测一个药物没有副作用、没有毒性,虽然数据测试正确率很高,但是如果不能说明为什么没有毒性,大部分人都会持有保守态度,可能还需要时间检验,因为只要有1%的差错率都会带来严重后果。另一方面,学术界关心如何把化合物更好地进行表征,即使图神经网络比之前的分子指纹方法要好很多,已经能表征一些结构信息,但是也损失了一些化合物的信息,因为它只表征了二维信息甚至局部信息。要把整个化合物的结构以及影响生物活性的属性更好的表征成为数字化的信息输入神经网络,需要学术界进一步的开发和研究,可能会进一步提高化学合成的的预测准确度。
(全文结束)